https://doi.org/10.1007/s10854-026-17395-6
(Accepted April 2026, Published May 2026)
Potassium doped TiO2 nanotube electrodes of different phases were synthesized by electrochemical anodization followed by doping, adopting two different strategies. The K-doped mixed-phase electrodes, prepared by an innovative ‘water bath temperature-controlled electrochemical anodization and doping method, exhibited specific capacitance values four to six times higher than those of the K-doped anatase electrodes prepared by the conventional electrochemical anodi zation, doping, and annealing. A maximum specific capacitance of 429 mF cm−2 (714 F g−1) at a current density of 0.75 mA cm−2 in a 1.23 V potential was obtained for the K-doped mixed-phase electrode, while the K-doped anatase yielded a specific capacitance of only 96 mF cm−2 (172 F g−1) at 3.8 mA cm−2. The former showed consistent performance in a wider potential window of 1.5 V, manifesting enhanced values of specific capacitance ~ 639 mF cm−2 (1065 F g−1) at 3.3 mA cm−2. The mixed-phase electrode manifested excellent cyclic stability retaining 84% of its initial capacitance value over 10,000 charge discharge cycles. An asymmetric supercapacitor, developed using the K-doped mixed-phase electrode and an activated carbon electrode as the negative and positive electrodes, respectively, operated in 2.1 V potential window, achieving a specific capacitance of 167 mF cm−2 (173 F g−1) at a scan rate of 5 mV s−1. The supercapacitor retained 83% of its initial specific capacitance value after 4800 cycles of operation. Three supercapacitors in series successfully powered a red LED of forward voltage of 1.8 V for 15 min.
https://doi.org/10.1002/est2.70416
(Accepted April 2026, Published May 2026)
This work explores the influence of two different fabrication strategies on the electrochemical performance of Fe doped TiO2 nanotube electrodes for supercapacitors. Electrodes of anatase phase (TNT/Fe-450) and mixed phases of TiO2 (B) and brookite (TNT/Fe-60) were prepared, respectively, by the conventional electrochemical anodization and a novel “water-bath temperature-controlled anodization” method, respectively. While the TNT/Fe-450 electrode exhibited a maximum areal (gravimetric) specific capacitance of only 86.48 mF cm−2 (166.30 F g−1), the TNT/Fe-60 showed nearly 10-fold enhancement in energy storage efficiency, with a maximum specific capacitance of 952.59 mF cm−2 (1693.49 F g−1) from cyclic voltammetry. Galvanostatic charge discharge measurements yielded a maximum specific capacitance (Cs) of 1427.99 F g−1, an energy density (ED) of 300 W h kg−1 and a power density (PD) of 2.43 kW kg−1, for TNT/Fe-60. Asymmetric supercapacitors of two configurations with TNT/Fe-60 as the negative electrode and either activated conducting carbon cloth (ACC) or MnO2 over CC, as the positive electrode were assembled. The MnO2-paired ASC demonstrated the best performance, with a high Cs of 352.19 Fg−1 at 10 mV s−1, ED of 94.05 W h kg−1, PD of 5.46 kW kg−1, and 100% capacity retention in 5 k cycles, indicating its potential for high energy storage applications.
https://pubs.acs.org/doi/10.1021/acs.langmuir.6c00202
(Accepted March 2026, Published March 2026)
A serious environmental threat is posed by the discharge of organic pollutants into water bodies, necessitating the development of sustainable remediation technologies. This paper discusses the effectiveness of bromine-treated titanium dioxide (TiO2) nanotubes, synthesized by a cost-effective and scalable electrochemical method, for the photocatalytic degradation of three highly toxic dyes: crystal violet (100% at pH 10) in 120 min and methylene blue (96.8% at pH 10) and methyl orange (98.6% at pH 3) within 180 min of irradiation. The photodegradation rate constants for the bromine-treated photocatalysts exhibited a remarkable increase compared to the untreated samples. The photocatalytic mechanism was systematically investigated through radical scavenger experiments and pH-dependent zeta potential analysis. Bromine treatment significantly modified the electronic structure of TiO2, leading to a reduction in the band gap from 3.00 to 2.48 eV, the formation of abundant oxygen vacancies, and the partial reduction of Ti4+ to Ti3+ and Ti2+, as confirmed by X-ray photoelectron spectroscopy. Structural, morphological, and optical properties were characterized by using X-ray diffraction, Raman spectroscopy, field emission scanning electron microscopy, transmission electron microscopy, and diffuse reflectance spectroscopy. Cyclic stability tests demonstrated the excellent reusability of the photocatalyst. These findings highlight bromine-treated TiO2 nanotubes as a promising and efficient photocatalyst for wastewater treatment and environmental remediation applications.

https://doi.org/10.1039/D6NJ00006A
(Accepted February 2026, Published March 2026)
The growing demand for sustainable, cost-effective, and high-performance energy storage technologies has intensified research into alternative systems beyond lithium-ion batteries (LIBs). Sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs) have emerged as promising candidates due to the abundance and affordability of Na and K. In this study, density functional theory (DFT) methods were employed to systematically investigate the potential of twenty-three polycyclic aromatic hydrocarbons (PAHs) as organic anode materials for SIBs and PIBs. A detailed analysis of the interaction between PAHs and alkali metal cations (M+ = Na+, K+) and atoms (M = Na, K) was performed using molecular electrostatic potential (MESP) analysis, adsorption energy calculations, charge transfer analysis, and spin density distribution analysis. The results reveal that the interaction between PAHs and M leads to charge transfer, resulting in the formation of PAH˙−⋯M+ complexes, which are crucial for anode activity. Cell voltages (Vcell) were computed from adsorption energetics, highlighting a strong dependence on the structure and size of the PAH. Compact and highly conjugated PAHs like naphthalene, phenanthrene, coronene, and circumbiphenyl show superior electrochemical performance, with several candidates demonstrating Vcell > 1.00 V for LIBs and SIBs and Vcell > 0.60 V for PIBs. This study establishes key structure–property relationships, underscoring the importance of molecular geometry, π-conjugation, and electron-donating ability in tailoring organic anode materials for post-lithium battery systems.

https://doi.org/10.1021/acs.inorgchem.5c05869
(Accepted February 2026, Published February 2026)
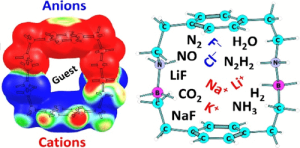
Azaboracyclophanes constitute a new class of three-dimensional, charge-separated molecular cavities in which B–N dative bonds function as internal polarizing elements, generating oriented electrostatic environments akin to confined electric fields. Using density-functional theory, we show that systematic incorporation of B–N linkages into cyclophane scaffolds produces large and programmable intramolecular polarity, shortens B–N distances, and amplifies internal MESP minima by up to ∼ 3× relative to carbon analogues. This electrostatic engineering profoundly reshapes host–guest behavior: azaboracyclophanes strongly stabilize cations and anions on both outer surfaces and within the cavity, convert a weakly binding parent framework into an ambivalent receptor, and enhance internal binding of polar guests by up to 57%. Distinct BN-placement motifs (NN7BB vs NB7BN) enable selectivity switching, including preferential stabilization of quadrupolar CO2. These results establish B–N-doped cyclophanes as programmable supramolecular “reaction chambers,” where electric-field-like effects emerge from molecular design rather than external application. The principles revealed here suggest general strategies for ion recognition, gas capture, and field-assisted reactivity within polarizable nanocavities.
https://doi.org/10.1021/acs.jpca.5c08587
(Accepted February 2026, Published February 2026)
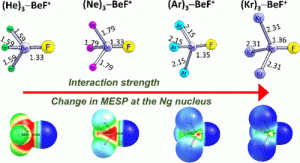
The interactions of noble gases (Ng = He, Ne, Ar, Kr) with highly electron-deficient main-group fragments are systematically investigated by using high-level CCSD and CCSD(T) calculations. A broad set of electrophilic acceptors is considered, spanning six-electron (O, S, F+, Cl+, Br+, OH+, SH+, NH2+), four-electron (BF2+, AlF2+), and two-electron (BeF+, MgF+) centers. Optimized geometries, interaction energies, and electronic descriptors reveal a continuous evolution of Ng binding behavior across the series from weak polarization-dominated interactions for He and Ne, to donor–acceptor bonding for Ar, and to strongly covalent-like coordination in Kr complexes. The analysis, supported by natural bond orbital (NBO), quantum theory of atoms in molecules (QTAIM), symmetry-adapted perturbation theory (SAPT), and molecular electrostatic potential (MESP) descriptors, demonstrates that the strength and multiplicity of Ng binding are governed primarily by the electrophilicity and valence-electron deficiency of the acceptor fragment with noble-gas polarizability modulating the interaction strength. Within this context, a unified 2e–4e–6e valence-electron framework is employed as a descriptive tool to rationalize why six-electron centers preferentially bind one Ng atom, four-electron centers stabilize two Ng atoms, and highly electron-deficient two-electron centers accommodate three Ng atoms. Among the multi-noble-gas complexes examined, BeF+ and MgF+ are found to stabilize tri-noble-gas adducts across the He–Kr series, with Kr3BeF+ exhibiting the strongest overall binding. Trihelium coordination to BeF+, with interaction energies of several kcal mol–1 per He atom, highlights the remarkable stabilization that can arise in extreme electron-deficient environments. Overall, the results provide a unified and internally consistent framework for organizing mono-, di-, and tri-noble-gas binding motifs across the noble-gas series, clarifying the electronic factors that govern noble-gas coordination in highly electrophilic chemical regimes.
https://doi.org/10.1039/D6OB00128A
(Accepted February 2026, Published February 2026)
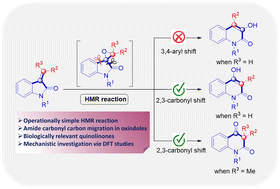
House–Meinwald rearrangement of oxindole substrates facilitating a one-pot synthesis of 3-substituted 4-hydroxyquinolin-2-ones and quinoline-2,4-diones under EBHP and base-mediated conditions is reported. This transformation proceeds through a well-defined sequence involving epoxidation, formation of a quinone methide-type intermediate, and subsequent ring expansion. The crucial carbonyl migration step was further substantiated by DFT studies. Moreover, the synthetic utility of the resulting 3-substituted 4-hydroxyquinolin-2-ones was demonstrated through late-stage diversification, highlighting the versatility of this protocol.
https://doi.org/10.1039/D5CP04586J
The cooperative interaction of nitrogen and oxygen centers in aromatic heterocycles provides an effective pathway for charge-assisted CO2 capture. Building on the pioneering work of Luo et al. on hydroxy-pyridine systems and our recent PCCP study on hydroxy-substituted N-heterocycles, this work extends the O,N-cooperative binding concept to triazolate frameworks. Density functional theory (DFT) and molecular electrostatic potential (MESP) analyses were performed on neutral, monoanionic, and dianionic 1,2,3- and 1,2,4-triazoles to elucidate how charge, counter ion and solvation govern CO2 adsorption. Deprotonation generates anionic and dianionic triazolates with enhanced negative potential at N and O sites, enabling O-carboxylate and N-carboxylate formation. While gas-phase dianions capture multiple CO2 molecules through cooperative charge delocalization and carbonate-chain growth (up to three CO2 with ΔGad ≈ -54 kcal mol-1), the inclusion of ethanol solvation and tetramethylphosphonium counter-ions reveals that these species remain strongly exergonic. Reaction modeling shows that both mono- and dianionic triazolates undergo spontaneous CO2 fixation in solution (ΔG ≈ -24 to -61 kcal mol-1), forming ion-paired poly(carboxylate) complexes with up to six CO2 molecules. These results demonstrate that even under polar, solvated conditions, counter-cation-stabilized triazolates preserve high-affinity, multi-site CO2 capture, identifying them as realistic and promising building blocks for molecular CO2-activation strategies.
https://doi.org/10.1007/s10800-025-02426-7
(Accepted October 2025, Published Dec 2025)
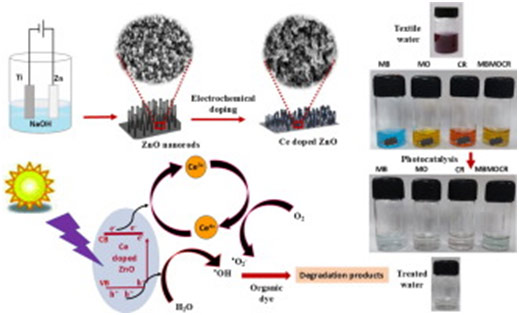
The improper management of industrial by-products has become a pressing global issue, posing serious risks to both environmental sustainability and public health. Safeguarding water bodies from harmful contaminants is imperative to preserve ecological balance and to ensure the well-being of life on Earth. The present study demonstrated a highly efficient approach for degrading toxic organic dyes – Methylene Blue, Methyl Orange, Congo Red, and their mixture, through photocatalysis using copper-incorporated zinc oxide nanostructures. The nanostructured photocatalysts were synthesized directly on zinc foil via a straightforward and scalable electrochemical anodization method. The research highlighted the crucial role of optimizing both the copper doping duration and the pH of dye solutions to enhance photocatalytic performance. The superior efficiency of copper-incorporated zinc oxide nanostructures was attributed to the Cu2+/Cu1⁺ redox couple, which acted as an effective electron trap, minimizing electron-hole recombination and promoting catalytic activity. Additionally, copper incorporation led to a notable reduction in the optical band gap, enhancing visible light absorption. The pH-dependent behaviour of the photocatalytic process was linked to the zeta potential of the catalyst and an optimal pH range was identified for the maximum degradation of each dye. Comprehensive structural, optical, compositional, electrochemical impedance and Mott-Schottky analyses further elucidated the mechanisms behind the improved performance. The work underscored the remarkable potential of anodized copper-incorporated zinc oxide nanostructures as highly efficient photocatalysts for the treatment of wastewater containing various dye pollutants, offering a promising pathway towards sustainable environmental remediation.

https://doi.org/10.1016/j.jpcs.2025.113425
(Accepted Nov 2025, Published Dec 2025)
Metal-doped mixed-phase TiO2 nanotube (TNTA) electrodes with enhanced performance characteristics for application in supercapacitors were developed by an innovative ”water-bath temperature controlled anodization” technique. The 4.9 % Mn-doped TNTA electrodes exhibited a high specific capacitance of 723.6 mF cm−2/1381.6 F g−1 at a current density of 6.3 mA cm−2, with a corresponding energy density of 276.3 Wh kg−1 and a power density of 7.2 kW kg−1, in a potential window of 1.2 V. Consistently high performance was observed across varying doping percentages and wider potential windows (PWs). An asymmetric supercapacitor with Mn-TNTA as the negative electrode and an activated conducting carbon cloth as the positive electrode achieved a specific capacitance of 192.7 F g−1, and energy and power densities of 91.5 Wh kg−1 and 1.1 kW kg−1, in a 2 V potential window. A superior asymmetric supercapacitor comprising MnO2-nanofibres modified TNTA as the positive electrode and Fe2O3-nanoparticles overlaid TNTA as the negative electrode, demonstrated a high specific capacitance of 286.6 mF cm−2/278.7 F g−1 and maximum energy and power densities of 170.7 Wh kg−1 and 1.9 kW kg−1 respectively. The device exhibited excellent cyclic stability over 5000 cycles with negligible performance degradation within a 2.1 V potential window. A single ASC powered a red LED (forward voltage: 1.8 V) for 9 min 30 s, while two ASCs in series powered the same red LED for 2 h 30 min and a violet LED (forward voltage: 3.7 V) for 1 h 9 min before complete dimming.

https://doi.org/10.1039/D5NJ02489G
The non-biodegradable nature of polyethylene terephthalate (PET) raises concerns about the accumulation of post-consumer PET products in the environment. Recycling is the most viable solution to address this issue. Among PET recycling methods, glycolysis has gained commercial interest due to favourable reaction conditions. Ionic liquids (IL) have emerged as promising green catalysts for PET glycolysis. This study highlights the use of 1-ethyl 3-methyl imidazolium dicyanamide ([EMIm][DCA]) ionic liquid as an efficient catalyst for PET degradation. Probable reaction mechanisms and side reactions encountered were also discussed. Under optimised reaction conditions, bis-hydroxy ethylene terephthalate (BHET) yield of 93% and 100% PET conversion was achieved. A BHET yield of over 57% was obtained in just 0.5 hours. Neutral hydrolysis of the ester bond was identified as a significant side reaction at high concentrations of [EMIm][DCA]. This process results in the formation of monohydroxy ethylene terephthalate (MHET), which is also valuable monomer for various synthesis applications. The possible side reactions and reaction products were identified and quantified by HPLC, NMR spectroscopy and chemical titrations. IL catalysed glycolysis reaction was found to follow the pseudo first order kinetics with an experimental activation energy of 168 kJ mol− 1. The mechanism of catalysis was explored through DFT studies, revealing the key role of hydrogen bonding interactions between IL and ethylene glycol (EG) in catalysing glycolysis reactions. DFT and spectroscopic methods confirmed the hydrogen bonding between EG and [EMIm][DCA] with a computed interaction energy of –118 kJmol⁻¹. Based on DFT studies a novel two-step mechanism was proposed for the glycolysis. Computed activation barrier for the catalysed glycolysis is 166 kJmol⁻¹ against 204 kJmol⁻¹ for the uncatalyzed.
https://doi.org/10.1007/s11224-025-02635-y
Molecular electrostatic potential (MESP) analysis has emerged as a powerful and chemically intuitive framework for visualizing and interpreting electronic structure features such as delocalization, aromaticity, and lone pair localization. This review highlights how the topography of the scalar field of MESP—defined through its minima and critical points (CPs), particularly (3, + 3) CPs—serves as a spatially resolved descriptor that captures subtle electronic phenomena often obscured in orbital-based or magnetic criteria. Applications to benzenoid hydrocarbons, linear acenes, heteroaromatics, and fused aromatic–antiaromatic hybrids illustrate how MESP reflects patterns of π-electron delocalization, including effects of curvature, strain, and fusion defects. In parallel, recent developments have shown that MESP topography can rigorously identify localized lone pairs based on the curvature and directionality of negative-valued CPs, offering a physically observable distinction from delocalized π-regions. These lone pair signatures have proven valuable in understanding noncovalent interactions such as hydrogen bonding, lone pair–π contacts, and coordination to electrophiles, with strong correlations between MESP minima and interaction energies. The review also emphasizes the unique advantage of MESP as a geometry-sensitive, scalar field approach that bridges empirical chemical intuition with quantum mechanical observables. Overall, MESP topography unifies the analysis of delocalized and localized electron distributions, providing a versatile platform for interpreting reactivity, guiding ligand design, and probing structure–function relationships in molecular systems.

https://doi.org/10.1039/D5NJ02780B
This study presents a comprehensive computational investigation of pristine and BN-doped polycyclic aromatic hydrocarbons (BN_PAHs) as potential anode materials for alkali metal-ion batteries (MIBs), including lithium-, sodium-, and potassium-ion systems. Using molecular electrostatic potential (MESP) analysis, Mulliken charge analysis, spin density distribution, and adsorption energy calculations, we examined the interactions of BN_PAHs with Li, Na, and K in both atomic and cationic forms. Results reveal that BN-doping enhances charge transfer and electron affinity, enabling stronger non-covalent interactions and tuning of adsorption strength. Charge transfer trends follow K > Na > Li for metal atoms and Li+ > Na+ > K+ for cations. A strong linear correlation (R > 0.97) between the MESP change at the metal nucleus (ΔVM+) and adsorption energy confirms ΔVM+ as a reliable descriptor of cation–π interactions. Cell voltage (Vcell) analysis identifies the (1BN_2⋯M) complex as the most promising anode material, delivering the highest voltages across systems: 1.41 V (LIB) and 0.81 V (PIB). These findings highlight the tunability and efficiency of BN-doped PAHs as next-generation organic anodes for MIBs.

https://doi.org/10.1007/s00339-025-08880-6
The removal of dyes from water, especially in wastewater treatment, is a critical environmental concern. This study demonstrates the efficient removal of dyes by adsorption and photocatalysis with Li-incorporated TiO2 nanotubes (Li-TNTAs) unannealed (~ 28 °C) and annealed at temperatures 250 °C and 600 °C. The annealing temperature significantly influences their structure, causing phase transitions and morphological shifts from tubular to interlocking structures, enhancing adsorption and photocatalytic performance. A dye removal of 99% in 10 min for Malachite Green dye by photocatalysis and 95.9% in 15 min by adsorption is achieved for Li-TNTA annealed at 600 °C. Kinetic and isothermal adsorption studies, along with pH effects (acidic, neutral and alkaline) in relation to the zeta potential of Li-TNTAs, are presented. Cyclic stability studies highlight the superiority of photocatalytic degradation over adsorptive removal during continuous use. For Li-TNTA annealed at 600 °C, the photocatalytic degradation efficiency remains stable over four consecutive cycles, whereas the adsorptive removal efficiency declines by 7.1% over the same number of cycles. Structural, morphological, compositional, and optical changes of Li-TNTAs, due to annealing are explored using various characterization techniques, with findings correlated to dye removal efficiency.
https://doi.org/10.1002/est2.70246
In this study, we report the effect of bromine treatment on electrochemically synthesized TiO2 nanotube (TNT) electrodes for supercapacitor applications. The bromine-treated TNT electrodes, exhibiting an anatase crystal structure, showed a sixfold enhancement in specific capacitance compared to untreated TNTs, achieving a high gravimetric specific capacitance of 330 F g−1 and an areal specific capacitance of 135.59 mF cm−2 at a current density of 2.5 mA cm−2. Furthermore, the electrodes demonstrated exceptional cycling stability, retaining 100% of their capacitance after 5200 charge–discharge cycles. This remarkable performance enhancement is attributed to the generation of oxygen vacancies within the anatase lattice upon bromine treatment, along with a significant reduction in charge transfer resistance at the electrode–electrolyte interface. The material properties of the electrodes were comprehensively characterized using x-ray photoelectron spectroscopy, transmission electron microscopy, field-emission scanning electron microscopy, x-ray diffraction, and Raman spectroscopy. In addition, a prototype symmetric supercapacitor fabricated using the bromine-treated electrodes delivered a specific capacitance of 36.3 F g−1 at a scan rate of 5 mV s−1, a power density of 3.69 kW kg−1 and a capacitance retention of 100% over 2000 cycles.
https://doi.org/10.1016/j.est.2025.117649
Nanoarchitectured TiO2 is a benchmark material for electrochemical energy storage. Modifications like doping, metal decoration, and hybridization have helped in enhancing its efficiency. The present study introduces self-doped TiO2 nanotube electrodes (TNTAs), fabricated through a cost-effective electrochemical anodization followed by reduction in a water bath. These TNTA electrodes constituted of a mixture of brookite and TiO2(B), achieve record-high performance among TiO2 electrodes when specific capacitance, energy density (ED), and power density (PD) are considered collectively. The electrode, self-doped for 5 s in 0.5 M Na2SO4 manifests an areal/gravimetric specific capacitance of 665.85 mFcm−2 /1459.26 Fg−1 at a scan rate of 10 mVs−1 and a maximum ED of 121.76 Whkg−1 and a PD of 4.57 kWkg−1. The electrode shows good cycling stability, retaining 103 % of their capacitance after 500 cycles, 98 % after 800 cycles, and 80 % after 1600 cycles. Extending the self-doping duration to 10s enhances the ED and PD to 197.86 Whkg−1 at 2.9 kWkg−1 whereas increasing the molarity of the doping solution to 1.0 M increases them to 240.09 Whkg−1 at 4.9 kWkg−1 respectively. The exceptional performance of these specially fabricated electrodes arises from the synergy of intercalative and surface pseudocapacitive charge storage, rapid ion diffusion, low interfacial resistance, and a high concentration of oxygen vacancies and Ti3+ species. A symmetric supercapacitor with the TNTA has achieved a remarkable areal/gravimetric specific capacitance of 417.97 mFcm−2 /916.27 Fg−1 at 1.3 mA cm−2 in a 2 V potential window and a high ED of 509 Whkg−1 at a PD of 2.9 kWkg−1.

https://doi.org/10.1039/D5CP01075F
Hydroxy-substituted aromatic N-heterocycles, including hydroxy pyridine (py), dihydroxy naphthyridines (nt), and trihydroxy pyridonaphthyridines (pn), have been investigated for their potential as CO2 adsorbents using density functional theory (DFT) calculations. Building on the pioneering work of Luo et al., who demonstrated exceptional CO2 capture capacities in pyridine-containing anion-functionalized ionic liquids, this study extends the exploration to a broader range of N-heterocycles. These N-heterocycles exhibit exceptional CO2 capture capabilities, driven by cooperative interactions between nitrogen and oxygen centres with CO2. The adsorption capacity increases with the number of nitrogen centres and hydroxy groups, with py, nt, and pn systems binding one, two, and three CO2 molecules, respectively. Notably, anionic N-heterocycles exhibit dramatically improved CO2 adsorption compared to their neutral counterparts, forming covalent bonds with CO2. The presence of counter cations, such as lithium or tetramethylphosphonium ions, further stabilizes CO2 adsorption, resulting in shorter interaction distances and higher exergonic free energy values. Solvent effects modeled using monoethanolamine (MEA) indicate a modest reduction in interaction energies for neutral and anionic systems, while ion-paired systems exhibit enhanced CO2 affinity in solution. Additionally, molecular electrostatic potential (MESP) analysis highlights the key adsorption sites and charge delocalization mechanisms that facilitate CO2 capture. The study also finds that enol–keto transformations, which could lead to CO2 conversion into carboxylates, are energetically unfavorable due to the loss of aromatic stability. These findings underscore the potential of hydroxy-substituted N-heterocycles, particularly in their anionic and cation-stabilized forms, as promising candidates for efficient CO2 capture. The insights gained from this study provide valuable guidelines for the design of next-generation CO2 sequestration materials and highlight new directions for experimental validation and real-world applications.

https://pubs.acs.org/doi/10.1021/acs.joc.5c00814
The Hammett substituent parameter (σₚ) is fundamental to physical organic chemistry but often suffers from inconsistencies in experimental determination across diverse substituents. This study presents a quantum chemical approach for estimating σₚ values using ¹H NMR chemical shift differences (Δδ) in hydrogen-bonded 2-pyridone heterodimers.
Using density functional theory (M06L/6-311++G(d,p)), we examined four dimer topologies—da, db, dc, and dd—and found that the Δδ values in the db configuration, where the substituent is spatially remote from the hydrogen-bonding region, exhibit the strongest correlation with σₚ. This observation aligns with the sterically free nature of σₚ.
Additional correlations were observed between σₚ and structural (Δr), energetic (ΔE), and electronic descriptors (MESP, QTAIM) of CO···HN hydrogen bonds. Among the topologies, db and dc systems provided the most reliable correlations, while deviations in da and dd dimers arose due to proximity-induced secondary interactions.
The NMR-derived substituent constant, σₚ(NMR), was computed for a diverse set of 99 substituents, offering a unified, reliable, and computationally efficient framework for predicting and refining Hammett constants. These findings underscore the potential of NMR chemical shifts as a powerful experimental and computational tool for quantifying substituent effects and enhance the theoretical foundation for structure–reactivity relationships in organic chemistry.

https://doi.org/10.1002/jcc.70122
Abstract
This study uses density functional theory (DFT) calculations at the M06-2X/6–311 + G(d,p) level to explore the potential of main group non-metals (X = O, N, S, P) to form hypervalent ylides (RmX+–YRn−2−). Substituent effects (R = alkoxy, alkyl, phenyl, H, NH2, NMe2, halogens) on the stability of these ylides and their neutral counterparts (Rm−1X–YRn−1) are analyzed across various X → Y bonding scenarios. Fluorine-substituted ylides (e.g., NhO/F, ShO/F, ShS/F, PhO/F) and PhO/OMe exhibited the highest stability compared to their neutral forms. Bond length and Wiberg bond order analyses reveal significant double bond character of the X–Y bond, confirming the hypervalent structure of the molecule. Molecular electrostatic potential (MESP) analysis shows reduced charge separation and delocalized electron density of the molecule, supporting a hypervalent ylene resonance form (RmX = YRn−2). These findings provide insights into the stability and electronic structure of hypervalent molecules, aiding the design of novel compounds.
https://doi.org/10.1021/acs.jpca.4c06558
Abstract
The phenomenon of positive cooperativity in noncovalent complexes, arising from electron donor–acceptor (eDA) interactions and subsequent electron reorganization, has been investigated by using density functional theory (DFT) at the ωB97XD/6–311 + G(3df,2pd) level. The study focuses on the interaction of various nitrogen- and oxygen-containing heterocycles with HF and HCl molecules. The formation of dimer complexes leads to electron flow from the nitrogen lone pair to the hydrogen halide, enhancing the electron density on the halogen atom, as made evident by molecular electrostatic potential (MESP) analysis. The introduction of additional HX molecules induces positive cooperativity, strengthening noncovalent N···H interaction and ultimately facilitating spontaneous H–X bond cleavage and formation of stable ion pairs. Substituent effects and positional isomerism in substituted pyridines reveal that electron-donating groups─especially at the ortho position─markedly enhance bond activation via neighboring group effects. Cooperative enhancement is also demonstrated in higher-order clusters (trimers to pentamers), particularly for the stronger H–F bond, which requires greater interaction synergy to cleave. The studies on O-heterocycles highlighted the impact of electronegativity on the extent of bond activation and the requirement for additional cooperative interactions to achieve H–X bond cleavage. The ΔVn(Cl) MESP parameter shows a strong correlation with interaction energy, serving as a predictive descriptor of bond activation. These findings provide valuable insights into the remarkable ability of weak noncovalent interactions to facilitate the breaking of strong bonds, offering insights with broad implications for catalysis, molecular design, and noncovalent bond activation strategies.
https://doi.org/10.1016/j.jpowsour.2025.237017
Abstract
The potential of TiO2 electrodes for supercapacitor applications is unlocked through a simple fabrication processthat has induced in-situ crystallization, with oxygen vacancy preservation, during anodization via a temperaturecontrolledwater bath for the electrolyte. The mixed-phase brookite-TiO2 (B) nanotube electrode fabricated at40°C delivers an exceptionally high energy density of 742 Whkg−1 without compromising power density (∼4.8kWkg−1) at a current density of 2.2 mAcm−2, across a wide potential window of 2V. The maximum areal specificcapacitance of the electrode ∼1382 mFcm−2 at 3.8 mAcm−2 is among the highest reported for TiO2 nanotubeelectrodes till date. The cyclic stability remains at 100 % for the first 9000 continuous cycles. A symmetric supercapacitorfabricated with the electrodes shows excellent performance delivering an energy density of 219Whkg−1 and a power density of 3.7 kWkg−1 at a current density of 1.7 mAcm−2. It yields a maximum areal capacitanceof 313 mFcm−2 or a gravimetric specific capacitance of 686 Fg-1 at a scan speed of 10 mVs−1. The remarkableperformance of this cost-effective and easily-reproducible electrode marks a breakthrough in the realm ofsupercapacitor electrode research, setting a new benchmark for future innovations.
https://doi.org/10.1007/JHEP04(2025)178
Abstract
The running coupling constant is calculated using the imaginary time formalism (ITF) of thermal field theory under the self-energy approximation. In the process, each Feynman diagram in thermal field theory is rewritten as the summation of non-thermal diagrams with coefficients that are functions of mass and temperature. By employing the same mass scale and coupling constant for both the non-thermal QFT and ITF, we derive a relation between them. Also, we calculate the self-energy using ITF, which is equated to the same as that of non-thermal QFT under the zero external momentum limit. This can provide a new expression for the coupling constant. Combining this result with the β(g) and γm(g) function relations of the renormalization group equations gives rise to a thermal-dependent coupling constant and running mass. Using these results, the free energy density is evaluated for two-loop order and compared with quasiparticle model.
https://doi.org/10.1140/epjp/s13360-025-06146-x
Abstract
We propose a modified liquid drop potential model to explore the thermodynamics of quark–gluon plasma (QGP) abovethe critical temperature, both in the absence and presence of amagnetic field. The model follows Mayer’s cluster expansion procedure.In this model, quarks and gluons are described as particles whose behavior is influenced by the QGP medium. Remarkably, evenat temperatures below the critical temperature (T < Tc), the equation of state for pressure and energy density across 2, 2+1 and 3flavors demonstrates a strong agreement with lattice data at zero magnetic field. Also, this modified liquid drop model is in goodagreement with lattice data for magnetized quark–gluon plasma in the region T > 113 MeV.
https://doi.org/10.1002/jcc.70078
Abstract
Ladderenes, unique polymers composed of fused cyclobutane rings, exhibit promising properties for energy storage applications, including reversible redox behavior and structural tunability. This density functional theory (DFT) study investigates the formation of ladderenes via gold(I)-catalyzed alkyne-alkene coupling reactions. Our calculations reveal a step-wise chain propagation mechanism where the alkene product of each step reacts with a ligand-gold-alkyne complex, leading to the sequential addition of cyclobutene units. We demonstrate that electron-withdrawing substituents on the alkynes, such as chlorine, facilitate the dissociation of the gold catalyst (PMe3Au+) from the cyclobutene adduct, enhancing catalytic efficiency. Solvent effects are also shown to significantly influence the reaction energetics. This finding highlights the significant impact of alkyne substitution on the catalytic cycle. The inherent exothermicity of ladderene formation, coupled with their unique structural features, suggests their potential for storing energy in the form of “covalently condensed acetylene.
https://doi.org/10.1007/s10854-025-14259-3
Abstract
Measures to save our water bodies from pollutants are crucial for the healthysustenance of life in this age of water scarcity. Photocatalysis is an effective toolfor the treatment of water polluted by various industrial effluents. Here, potassium(K) doping is done to engineer the structural, morphological and opticalproperties of TiO2nanotube arrays for the improvement of its photocatalyticdegradation efficiency. Doping enhanced the photocatalytic degradation ratesto ~ 96% in 120 min and ~ 97% in 180 min, respectively, for crystal violet andmethylene blue dyes from the respective values of 70% and 55% for pristine TiO2nanotubes. The fabrication and doping of the nanotube arrays vertically alignedover titanium foil are done by the cost-effective electrochemical method. Frommultiple characterizations, the enhancement in the photocatalytic efficiency of thedoped nanotube arrays is ascribed mainly to the drastic reduction in the band gapfrom UV to the visible range, formation of carrier trap centers by reduction of Ti4+to Ti3+/Ti2+ states, presence of more hydroxyl groups, an increased orientation ofK+doped nano-crystallites along (004) plane.
https://doi.org/10.1016/j.est.2025.115289
Abstract
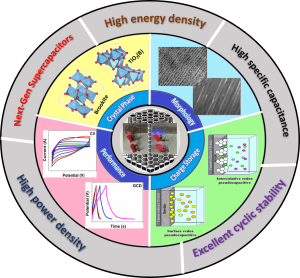
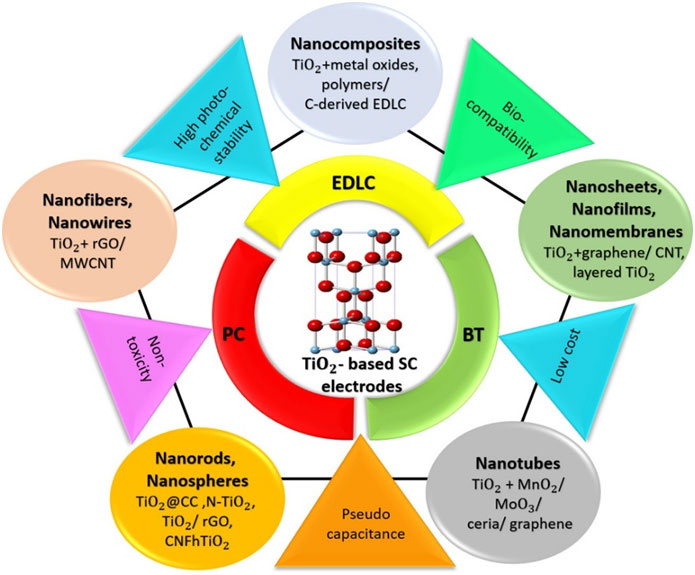
This review provides a comprehensive overview of recent advancements in TiO₂-based nanomaterials as electrodesfor supercapacitors, emphasizing strategies that enhance their electrochemical performance. It details thesynthesis methods and modification techniques such as hybridization with carbon-based materials, metal iondoping, and combination with pseudocapacitive materials of various TiO₂nanoarchitectures, including 0-Dnanoparticles, 1-D nanowires, nanorods, nanofibers, nanotubes, 2-D nanosheets, and 3-D nanocomposites forsupercapacitor electrodes. The impact of these modifications on key performance parameters, such as specificcapacitance, energy and power densities, and cycle life, is critically evaluated. Notably, we highlight the significantimprovements in energy density achieved through the aforementioned modifications, reaching values of~102 Wh kg-1at power densities nearing 104 W kg-1. The review also addresses ambiguities in the interpretationof charge storage mechanisms, providing clarity on electric double-layer capacitance, pseudocapacitance,and battery-type intercalation, and critically examines inconsistencies in their classification and analysis in theliterature. It categorizes supercapacitors based on their electrode types, distinguishing between symmetric,asymmetric, and hybrid configurations, and underscores the performance impact of different charge storageprocesses. The review concludes by outlining future research directions to further optimize TiO₂-based electrodeperformance to facilitate their widespread application in high-performance energy storage devices.
https://doi.org/10.1039/D4NJ05047A
Abstract: 1-Ethyl-3-methylimidazolium dinitramide (EMImDN), an energetic ionic liquid, was synthesized and characterized by IR, 1H NMR, 13C NMR, 15N NMR, and 17O NMR techniques. The thermal properties of EMImDN were investigated through DSC and TG analyses. The TG curve revealed three consecutive exothermic decomposition events with peak temperatures at 223 oC, 277 oC and 308 oC, accompanied by an 80 % mass loss between 180 oC and 280 oC. The thermal decomposition kinetics were analyzed using the Kissinger method and Flynn-Wall-Ozawa isoconversional method. The activation energies for the three decomposition stages were calculated to be 41.1, 35.9 and 43.2 kcal/mol respectively by Kissinger method and 39.0, 36.8 and 42.6 kcal/mol respectively by FWO method. Pyrolysis GC-MS analysis identified the decomposition products, and the decomposition mechanism was predicted to involve dealkylation of the imidazole ring via bimolecular nucleophilic substitution (SN2). The proposed decomposition mechanism was further supported by density functional theory (DFT) calculations at B3LYP/6-311+G(d,p) level. Additionally, the molar enthalpy of formation of EMImDN (+54.5 kcal/mol), determined through combustion calorimetry, underscores the energetic nature of this ionic liquid. Notably, a 10 wt.% solution of NH3BH3 in EMImDN exhibited hypergolicity with red fuming nitric acid. Preliminary investigations into the interactions of EMImDN with HNO3 and NH3BH3 provide insights into the initial stages of this hypergolic reaction.
https://doi.org/10.1039/d4dt02703e
Abstract
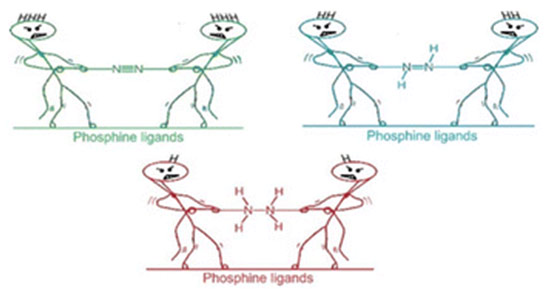
Activating atmospheric dinitrogen (N2), a molecule with a remarkably strong triple bond, remains a major challenge inchemistry. This theoretical study explores the potential of superbase phosphines, specifically those decorated withimidazolin-2-imine ((ImN)3P) and imidazolin-2-methylidene ((ImCH)3P) to facilitate N2 activation and subsequent hydrazine(H2NNH2) formation. Using density functional theory (DFT) at the M06L/6-311++G(d,p) level, we investigated the interactionsbetween these phosphines and N2. Mono-phosphine-N2 complexes exhibit weak, noncovalent interactions (-0.6 to -7.1kcal/mol). Notably, two superbasic phosphines also form high-energy hypervalent complexes with N2, albeit at significantlyhigher energies. The superbasic nature and potential for hypervalency of these phosphines lead to substantial N2 activationin bis-phosphine-N2 complexes, where N2 is “sandwiched” between two phosphine moieties through hypervalent P-N bonds.Among the phosphines studied, only (ImN)3P forms an exothermic sandwich complex with N2, stabilized by hydrogenbonding between the ImN- substituents and the central N2 molecule. A two-step, exothermic hydrogen transfer pathwayfrom (ImN)3P to N2 results in the formation of a bis-phosphine-diimine (HNNH) sandwich complex. Subsequent hydrogentransfers lead to the formation of a bis-phosphine-hydrazine (H2NNH2) complex, a process that, although endothermic,exhibits surmountable activation barriers. The relatively low energy requirements for this overall transformation suggest itspotential feasibility under optimized conditions. This theoretical exploration highlights the promise of superbase phosphinesas a strategy for metal-free N2 activation, opening doors for the development of more efficient and sustainable nitrogenfixation and utilization methods.
https://doi.org/10.1016/j.jiec.2024.11.025
Abstract
The development of cost-effective water-treatment methods to remove toxic industrial contaminants is of burgeoning demand today. This paper discusses the superior performance of Ce doped ZnO for the photocatalytic degradation of toxic cationic and anionic dyes. The excellent degradation ability of this easily manoeuvrable solid photocatalyst developed by the facile and low-cost electrochemical method, for the simultaneous removal of multiple dyes, avoiding the need for any post treatments is successfully demonstrated with the synthetic dyes methylene blue, methyl orange, congo red and their mixture. The pH dependence of the photocatalytic efficiency is correlated with its zeta potential. The improved photocatalytic efficiency is attributed to the Ce4+↔Ce3+ redox couple acting as trap centers enhancing the production of superoxide radicals and the reduction of carrier recombination. The scavenger and cyclic stability tests confirm respectively the dominant role of superoxide radicals in photocatalysis and the reusability of the photocatalyst. The photocatalyst is characterized in detail by X-ray diffraction, X-ray photoelectron spectroscopy, field emission scanning electron microscopy, Rutherford backscattering, high-resolution transmission electron microscopy, selected area electron diffraction, surface charge analysis and diffuse reflectance Spectroscopy. The study highlights the prospect of Ce doped ZnO nanostructured film as a high-performing photocatalyst for water treatment.
https://doi.org/10.1002/pssa.202400612
Abstract
A highly efficient, solid, and easily maneuverable adsorbent and photocatalyst, Li-incorporated titanate nanotubes that manifest adsorption efficiency of 98.1% and photocatalytic efficiency of 99.6% in 60 min and 97.8% and 98.5%, respectively, in a shorter duration of 20 min, is synthesized by a facile two-stage electrochemical technique. The modified nanotubes exhibit good cyclic stability of 93.7% photodegradation of malachite green dye after three continuous cycles. The adsorption data show the highest correlation for second-order pseudo kinetics and Freundlich isotherm, suggesting multilayer chemisorption with an adsorption capacity of kF ≈ 219.51 mg1–1/nL1/ng−1. Fourier transform infrared spectroscopy (FTIR) analysis of the adsorbent before and after the adsorption corroborates the findings. X-Ray photoelectron spectroscopy reveals the formation of a larger number of oxygen vacancies (Ti3+/Ti2+) that facilitate more carrier release for the production of reactive oxygen species during photocatalysis. X-Ray diffraction and transmission electron microscopy analysis confirm a secondary rutile phase formation on Li doping that promotes heterogeneous multilayer adsorption. Field-emission spectroscopic studies reveal a change in the morphology of the doped tubes with porous aggregate formation on the surface. The structural, morphological, and compositional change brought about by Li incorporation facilitates the use of titania nanotubes as an efficient adsorbent cum photocatalyst in the removal of malachite green dye.
https://doi.org/10.1002/wcms.1735
Abstract
Molecular electrostatic potential (MESP) analysis has emerged as a pivotal tool in drug discovery, providing insights into molecular reactivity and noncovalent interactions essential for drug function. While widely used MESP-on-isodensity surface analysis offers interpretations of electron-rich or deficient regions of a drug molecule, the MESP topology parameters such as spatial minimum (Vmin) and MESP at nuclei (Vn) provide a quantitative understanding. The investigation into the correlation between MESP parameters and various molecular properties such as lipophilicity, pKa (acidity/basicity), conformations, and tautomeric forms is crucial for understanding the impact on biological activity of drugs and facilitating drug design. Moreover, MESP topology analysis serves as a fundamental tool in elucidating the pharmacological behavior of compounds and optimizing their therapeutic efficacy. A quantitative study utilizing Vn parameters to assess the hydrogen bond propensity of a drug presents a novel strategy for investigating drug-receptor interactions with increased precision. The qualitative and quantitative analysis of the MESP features of various drugs, including their applications in cancer, tuberculosis, tumors, inflammation, and infectious diseases such as malaria, bacterial infections, fungal infections, and viral infections, is conducted in this review.
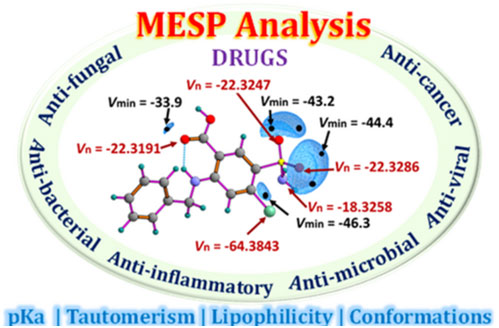
https://doi.org/10.1021/acsaom.4c00330.
Abstract
Two large-flexible porphyrinoids [40]pentathiadecaphyrin(1.0.1.0.1.0.1.0.1.0) S5N5 and [48]dodecaphyrin (1.0.1.0.1.0.1.0.1.0.1.0) S6N6 were obtained through Lewis acid catalyzed condensation of thiophene containing diheterole. The single crystal X-ray structure of S6N6 revealed a twisted “figure eight” conformation whereas the optimized structure of S5N5 displayed a coplanar arrangement of thiophene and pyrrole rings. Various spectral and theoretical studies along with the photophysical investigation of the SnNn (n = 3–6) series suggested that the higher order systems (S5N5 and S6N6) were deemed to be nonaromatic due to their nonplanar conformations. The transient absorption studies revealed a strong dependence on the electronic structure with conformational flexibility due to the expansion of the macrocyclic core. The internal conversion processes become significantly fast in higher order macrocycles SnNn (n = 5–6). These macrocycles are also shown to be promising candidates for nonlinear optical materials.
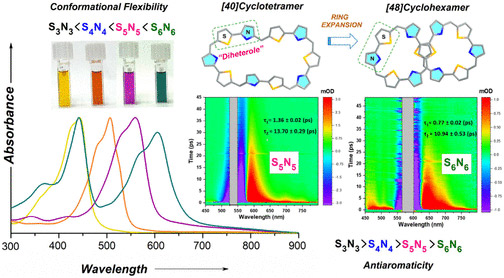
https://doi.org/10.1021/acs.joc.4c01635
Abstract
Members of a new class of bifunctional amino quaternary phosphonium salts have been synthesized and utilized as catalysts in aldol condensation reactions, as demonstrated herein. These secondary amines feature a phosphonium ion connected by a carbon chain, enabling the quaternary phosphonium ion to engage in distinct cooperative noncovalent interactions. These interactions work in tandem to stabilize different transition state complexes, exclusively controlling competing amine-catalyzed aldol pathways via the Mannich mechanism. Comprehensive mechanistic investigations were conducted through theoretical calculations. This study uncovers a proximity-driven catalytic mechanism in which the distance between the N and the P+ of the bifunctional catalyst emerges as a critical factor determining catalytic efficacy. The method has been demonstrated through its application to the total synthesis of several bioactive natural products.
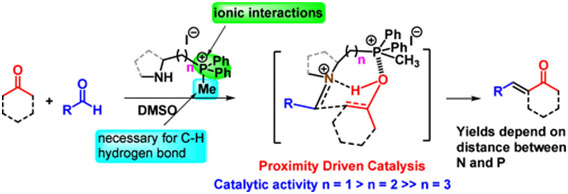
https://doi.org/10.1002/ejoc.202400450.
Abstract
An efficient method for the conversion of biphenyl acrylamides to dibenzoazepinones with −SCF3 incorporation is described. This operationally simple radical cascade reaction employs CAN as an oxidant and exhibits good functional group tolerance. Substrates featuring −OCH3, −CH3, −Br or −Cl at the para-position of the aromatic ring exhibits a preference for an ipso-cyclization due to the intervention of DMSO in the reaction. Density functional theory (DFT) calculations provide valuable insights into the reaction’s energetics and product selectivity.
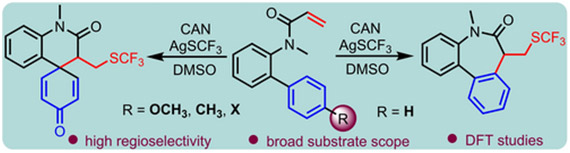
https://doi.org/10.1088/1361-6528/ad3649.
Abstract
The removal of pollutants from water bodies is crucial for the well-being of humanity and is a topic of global research. Researchers have turned their attention to green synthesized nanoparticles for wastewater treatment due to their eco-friendly nature, biocompatibility, and cost-effectiveness. This work demonstrates the efficient removal of organic dye and both gram-positive and gram-negative bacteria from water bodies using copper-doped cerium oxide nanoparticles synthesized with MurrayaKoenigii extract. Characterized via various methods, the 15% copper doped cerium oxide nanoparticles (Cu 15% NPs) exhibited maximum Congo red dye adsorption (98% degradation in 35 min). Kinetic analysis favoured a pseudo-second-order model, indicating the chemical nature of adsorption. Equilibrium adsorption isotherms aligned with the Langmuir model, indicating homogenous monolayer dye adsorption on the doped adsorbent. The maximum uptake of adsorbate, Qm obtained from Langmuir model for Cu 15% NPs was 193 mg g−1. The study also showed enhanced antibacterial activity against Bacillus subtilis, Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa for Cu-doped ceria, attributed to generation of reactive oxygen species (ROS) induced by the redox cycling between Ce3+ and Ce4+. This substantiated that the green synthesized copper doped cerium oxide nanoparticles are potential candidates for adsorptive removal of Congo red dye and as antibacterial agents.
https://doi.org/10.1039/D4CP03371J
Abstract
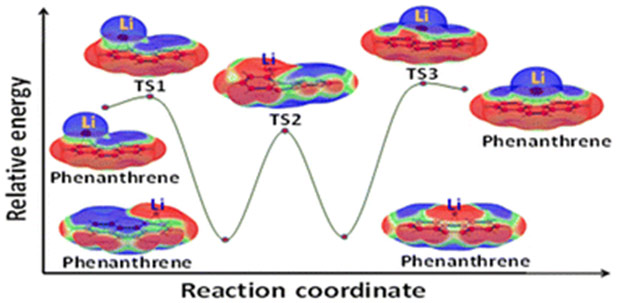
The study investigates the phenomenon of bond stretch isomerism (BSI) in complexes formed between alkali metals (Li, Na, K) and various non-aromatic, aromatic hydrocarbons, as well as heteroaromatic systems. The research employs density functional theory (DFT) calculations to optimize complex geometries and analyze their electronic structures using molecular electrostatic potential (MESP), charge, and spin density analyses. The results reveal that these complexes can exist in two distinct configurations: ‘loose’ long-bond isomers (lbi) that retain the original hydrocarbon geometry and ‘tight’ short-bond isomers (sbi) that undergo geometrical distortion upon complexation, with sbi generally being more stable. The interconversion between lbi and sbi occurs through a transition state. The study highlights the crucial role of electron transfer in BSI, with sbi involving valence electron transfer from the metal to the hydrocarbon, leading to zwitterionic radical complexes. In contrast, lbi exhibit a slight electron density transfer from the hydrocarbon to the metal. The presence of low-energy transition states between lbi and sbi suggests a dynamic shuttling mechanism for alkali metals, particularly Li, on hydrocarbon surfaces. The study identifies Li complexes as potential candidates for anode materials in batteries due to their stability and electron transfer properties, offering valuable insights into the design of advanced materials for energy storage applications.